1. 引言 :短肽药物设计——从“易降解”到“稳如磐石”
在药物研发的广阔版图中, 短肽 正以其独特的“中等身材”赢得越来越多的关注。相较于传统小分子药物,短肽能够形成更丰富的氢键和疏水相互作用,通常具有更高的结合特异性和亲和力,且代谢产物毒性较低;相较于抗体等大分子生物药,短肽则展现出更好的组织渗透性、更低的生产成本以及更弱的免疫原性。可以说,短肽在“小”与“大”之间找到了一个颇具吸引力的平衡点。
然而,天然短肽(由L-氨基酸组成)面临一个“天生短板”—— 体内蛋白酶降解 。人体内的蛋白酶能迅速识别并切割L-型肽链,导致许多短肽候选物的体内半衰期仅有几分钟到几十分钟。这一问题长期制约着短肽药物的临床转化。
一个优雅的解决方案是:将L-氨基酸替换为其手性对映体—— D-氨基酸 。D-氨基酸并非天然蛋白质的构成单元,因此能够有效逃逸蛋白酶的识别与降解,使短肽的代谢半衰期从分钟级延长至小时甚至天级。不仅如此,手性翻转还能解锁L-型肽无法进入的构象空间,为发现全新的结合模式提供了可能。正是这种“反手性”设计,让D-氨基酸短肽成为近年来的研究热点之一。
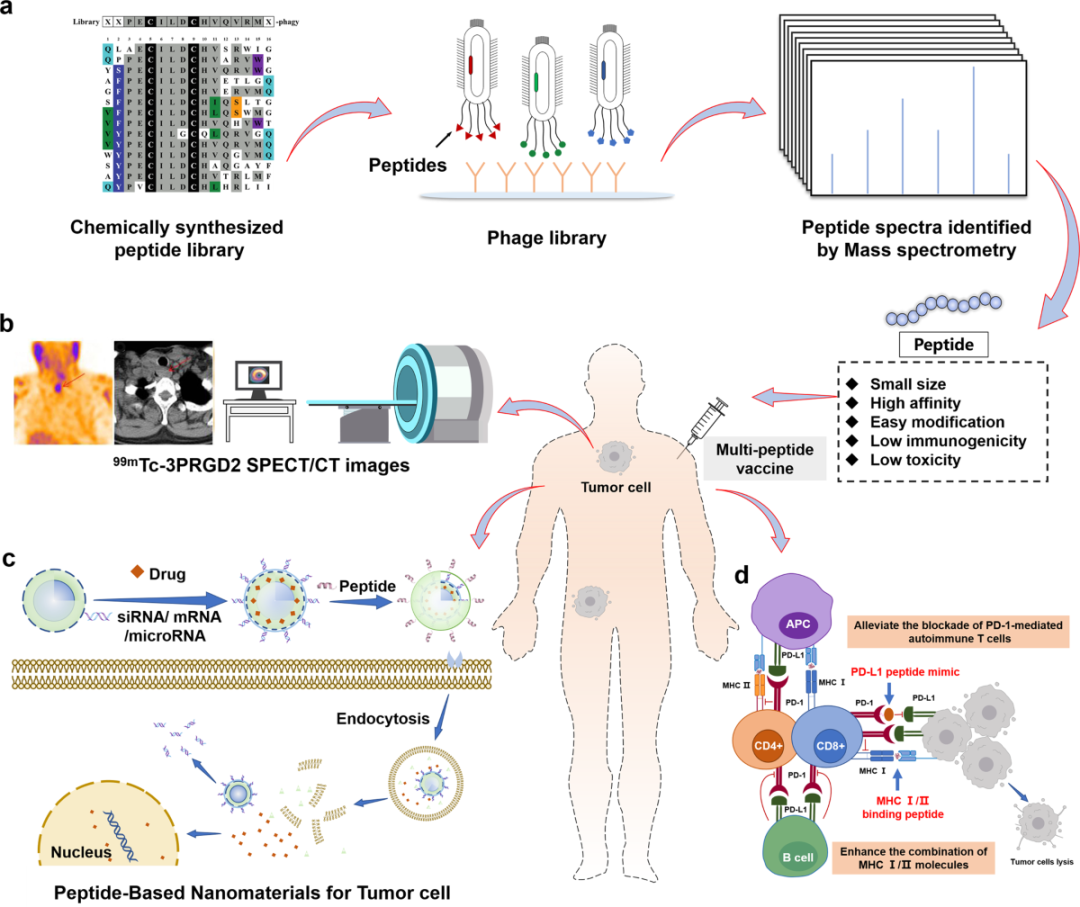
图1. 基于传统模式的短肽药物设计流程
2. 现代结构预测工具 :AI时代的新范式
传统上,短肽-蛋白复合物结构的获取高度依赖实验方法(如NMR、X射线晶体学)或分子对接。前者耗时费力,后者在面对柔性短肽时准确率往往不尽如人意。随着以 AlphaFold3 和 Chai-1 为代表的共折叠(co-folding)深度学习模型的问世,这一局面被彻底改写。
这些模型不再局限于单一蛋白质的折叠预测,而是能够直接预测 蛋白质-配体复合物 ,涵盖小分子、核酸以及短肽等多种配体类型。对于短肽设计而言,这意味着我们可以快速获得高置信度的结合模式假设,从而将“序列到结构”的周期从数周压缩到几十分钟。
但需要保持一份清醒的谨慎 。近期两项基准研究(Lai Luhua团队的工作Benchmarking co-folding methods to predict the structures of covalent protein–ligand complexes(https://doi.org/10.1038/s41401-025-01721-5),以及发表于*Nature Communications*的对抗性测试Investigating whether deep learning models for co-folding learn the physics of protein-ligand interactions(https://doi.org/10.1038/s41467-025-63947-5))系统评估了当前主流共折叠模型的物理合理性。结果显示,尽管这些模型在标准测试集上表现优异,但当面对结合位点被突变破坏、配体电荷或氢键能力发生剧烈改变等“对抗性”场景时,模型普遍表现出对原始结合位点的强烈偏倚——即使物理上已不应再结合,模型仍倾向于将配体“塞”回原处,并产生原子重叠、立体冲突等非物理结构。更令人担忧的是,高置信度指标(如pLDDT)往往无法反映这些错误。换句话说,模型可能在“自信满满”地输出一个物理上不可能的结构。
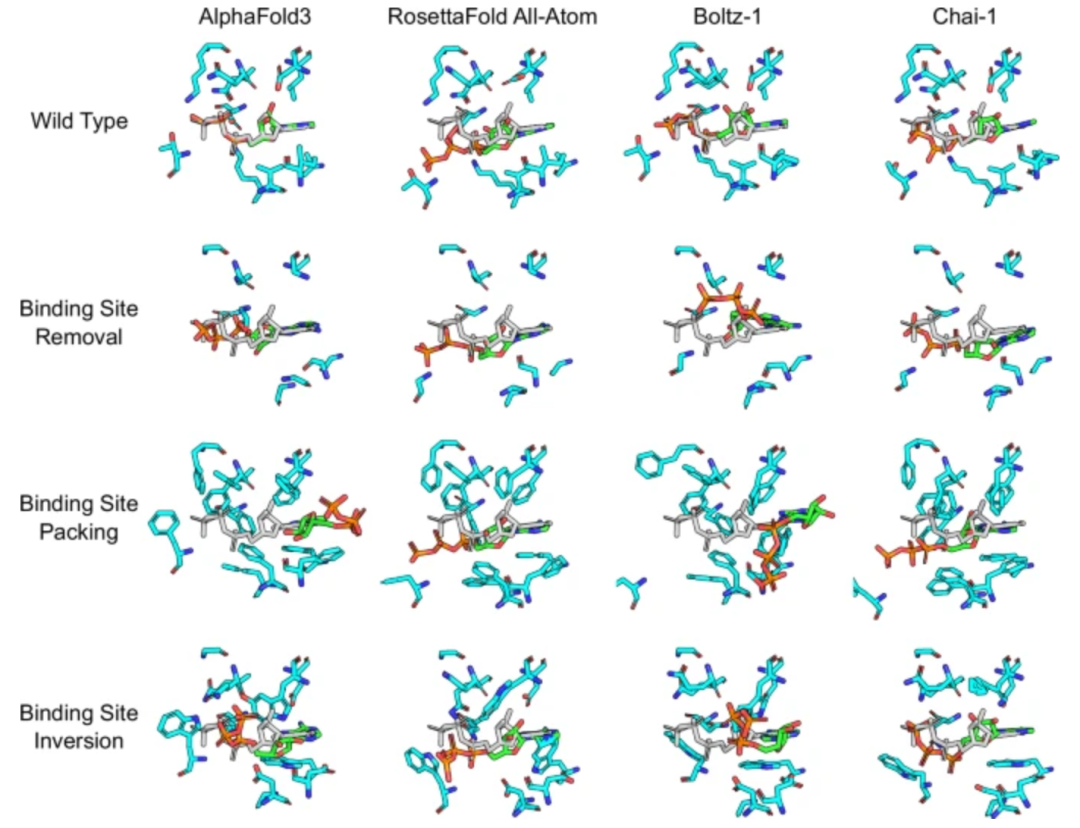
图2. 不同深度学习方法比较难对口袋修饰后的蛋白质-ATP结合模式做出准确预测
因此,研究者应当将AI预测视为“高效但偶有失误的初筛工具”,而非最终答案。 物理验证 ——尤其是分子动力学(MD)模拟——是弥补AI模型物理理解不足的必要环节。下文将介绍一套完整的“AI预测 + 结构映射 + MD模拟”工作流,专为D-氨基酸短肽设计量身打造。
3. D-氨基酸短肽的“预测+模拟”全流程方案
大多数AI模型(包括AlphaFold3)在处理D-氨基酸时,默认将其视为“非标准小分子配体”而非“蛋白质残基”。这会导致后续MD模拟面临原子命名混乱、缺乏标准力场拓扑等问题。我们搭建的自动化流水线打通了“ 文件准备 → AI预测 → 结构纠正 → MD模拟 ”的闭环,让格式兼容不再是障碍。
第一步:序列转SMILES——精确控制手性
对于D-肽,AI模型通常要求以 SMILES 字符串 形式作为“小分子”输入。最关键的一步是精确标记每一个α-碳的手性。可以考虑的工作流通过以下逻辑实现高精度SMILES生成:
-
LEaP构建
-
手性 翻转
-
拓扑转换
-
自动 手性 标记
# 示例:生成 D-His-D-Lys-D-His-D-Ala-D-Pro-D-Ser 的 SMILESpython scripts/d_peptide_smiles.py "NH2-D-His-D-Lys-D-His-D-Ala-D-Pro-D-Ser-Ac" -o ./prep_dir
第二步:自动化结构预测——一键调用
通过JSON配置文件定义靶点序列和短肽信息,即可一键调用AlphaFold3、Boltz-1或Chai-1进行预测。建议对同一体系进行多次重复预测(不同随机种子),以评估结构收敛性。
{"targets": [{"name": "target", "chain": "A", "sequence": "..."}],"peptides": [{"name": "peptide", "chain": "B", "sequence": "NH2-D-His-D-Lys-D-His-D-Ala-D-Pro-D-Ser-Ac", "is_d_amino_acid": true}],"predictions": [{"method": "af3", "run_script": "./run_af3.sh"}]}
第三步:结构纠正与“重肽化”——让格式归位
AI输出的CIF/PDB文件中,D-肽通常被标记为“小分子LIG”,原子名(C1, N1…)和残基名完全不符合MD模拟标准(AMBER力场要求N, CA, C, O等标准命名)。我们开发了 图匹配纠正算法 ,步骤如下:
-
拓扑感知
-
子图 同构 匹配
-
坐标映射
python scripts/map_af3_to_tleap.py -t template.pdb -i predicted.cif -o corrected_peptide.pdb -c complex.pdb第四步:修饰氨基酸与特殊基团的处理
许多D-肽设计中会引入非天然氨基酸或化学修饰(如磷酸化、甲基化、PEG化、荧光标记等)。对于这些“非标准”部分,标准的AMBER力场(如ff14SB)无法直接提供参数。此时需要采用 混合建模策略 :
-
标准骨架部分 :如果修饰基团连接在标准氨基酸侧链上(例如赖氨酸的侧链氨基上连接一个乙酰基),可以将该氨基酸主体保留为标准残基,仅对额外基团使用通用力场(GAFF2)生成参数。
-
完全非天然氨基酸 :若整个残基在标准库中不存在,则需要将其拆解为“类似的标准残基 + 修饰基团”,或从头使用GAFF2生成全参数,并通过原子类型、电荷和键参数的匹配确保与周围环境的兼容性。
-
连接处理 :利用
tleap的bond命令或第三方工具(如acpype、antechamber)将修饰基团共价连接到标准残基上,同时检查化合价和电荷的合理性。
具体操作上,可以先用
antechamber
生成修饰基团的GAFF力场参数,再用
resp
或
gasteiger
方法计算电荷,最后通过
tleap
将修饰基团与标准残基拼接成一个完整的残基。这一步需要仔细检查原子命名和连接顺序,避免“断键”或“错位”。对于常见的修饰(如磷酸化丝氨酸/苏氨酸/酪氨酸),AMBER力场已提供现成参数(如
SER
的磷酸化版本
SEP
),可直接调用。
第五步:分子动力学模拟的工具选择与能量计算的意义
模拟工具选择
目前主流的分子动力学模拟软件包括
AMBER
、
GROMACS
、
NAMD
、
OpenMM
等。对于短肽-蛋白复合物体系,建议优先考虑
AMBER
(力场参数丰富,尤其适合蛋白质-肽体系)或
GROMACS
(计算速度快,社区活跃)。若涉及复杂的自定义残基或修饰基团,AMBER的
tleap
配合
parmchk2
和
antechamber
提供了相对完整的解决方案。OpenMM则适合GPU加速和与机器学习势函数结合。选择时需综合考虑团队熟悉程度、力场兼容性以及是否需要对结合自由能进行后续计算(如AMBER的MMPBSA.py更为成熟)。本项目建议使用OpenMM更合适,可以支持大规模、自动化的模拟场景,方便进行肽设计和筛选。
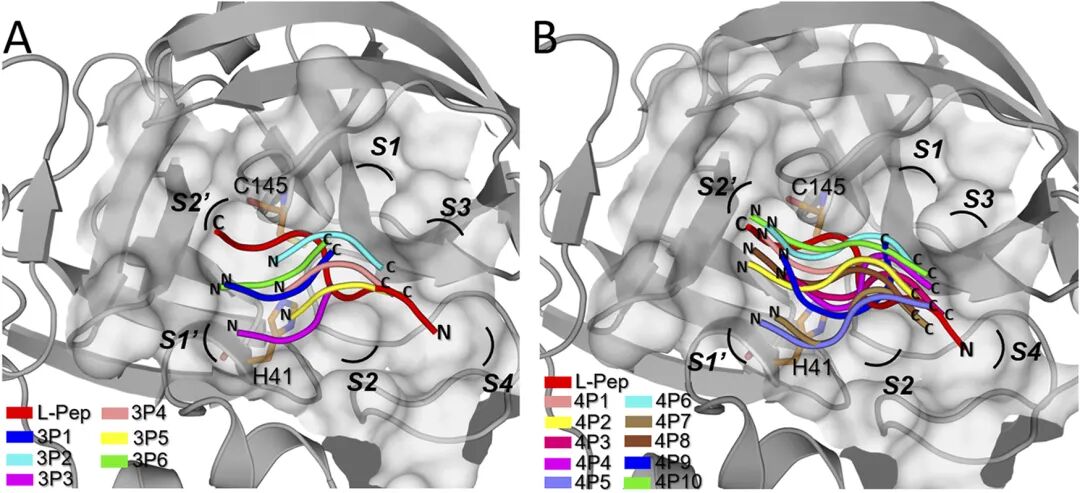
图3.基于分子东西模拟获得D-peptide和SARS-COV2主蛋白酶的结合模式
能量计算的意义
分子动力学模拟不仅仅是“看动画”。通过分析模拟轨迹中的能量项,可以量化设计的优劣:
-
势能组分分析 :分离键能、角能、二面角能、范德华能和静电相互作用能,帮助判断短肽与靶点之间的主要驱动力。
-
结合自由能(MMPBSA/MMGBSA) :这是评估亲和力的核心指标。通过计算蛋白-肽复合物与单独蛋白、单独肽之间的自由能差,可以得到结合自由能(ΔG)。该方法能够对不同序列、不同修饰的短肽进行相对排序,指导先导化合物优化。
-
残基贡献分解 :将结合自由能分解到每个残基上,识别“热点残基”——即对结合贡献最大的关键位置。这些热点往往是突变或修饰的重点关注对象。
-
熵 的估算 :通过正则模式分析或准谐波分析估算结合熵变,帮助理解结合过程中的构象柔性代价。
需要强调的是,能量计算的绝对值高度依赖力场参数和模拟设置, 相对比较 (如同一靶点下不同短肽的ΔG差值)比绝对数值更有意义。建议始终与阳性对照分子在完全相同的计算条件下进行比较。
能量计算的多元策略
MMPBSA/MMGBSA是评估短肽-蛋白结合亲和力的常用起点,因其计算成本相对较低且能提供残基能量分解,在序列筛选中具有实用价值。但研究者应认识到,MMPBSA对构象采样的质量高度敏感,其能量绝对值受力场参数和介电常数设置的影响较大,因此更适合用于 相对比较 (如同一位点下不同短肽设计的排序),而非作为绝对亲和力的可靠预测。
在MMPBSA的基础上,针对不同研究场景,可以考虑以下三种其他的能量计算策略:
① 深度学习 亲和力预测——高通量初筛的 加速器
近年来,面向蛋白-肽相互作用的深度学习亲和力预测模型发展迅速。例如,华中科技大学黄胜友团队提出的GraphPep框架,通过构建蛋白-肽相互作用图并利用层次图神经网络建模界面几何与能量特征,在多个基准数据集上的结合能预测与构象评分任务中均显著优于传统打分函数。类似地,PepBAN框架采用蛋白语言模型与双线性注意力网络,可有效学习肽-蛋白对的局部相互作用模式,并在环肽等非标准氨基酸体系中也表现出预测优势。此外,MMPepPro通过整合宏观结合趋势与微观残基相互作用特征,在近两万个肽-蛋白复合物上训练,展现了良好的泛化性能。
这些深度学习方法的共同优势在于 推理速度极快 (毫秒至秒级),适用于对数千条候选短肽进行结合倾向性的快速排序。但需要注意,这类模型的预测可靠性高度依赖于训练集的覆盖范围——当设计的短肽包含非天然氨基酸、D-型构型或新颖化学修饰时,模型可能外推失效。因此,深度学习亲和力预测更适合作为 高通量筛选 的第一道 过滤器 ,其输出需要结合物理模拟进行后续验证。但是这些深度学习方法,可能会饱受过拟合风险的困扰,让生物计算专家很难真正相信这些工具。
② 绝对结合自由能计算(ABFE)——无参考配体时的“从头”评估
当针对一个靶点没有任何已知活性分子作为参考时,深度学习模型无法提供可靠的相对排序,MMPBSA的绝对数值又缺乏可信度。此时, 绝对结合自由能计算(Absolute Binding Free Energy, ABFE) 成为一种更严谨的选择。ABFE通过构建热力学循环,将配体从结合口袋中“解耦”(逐步关闭其与蛋白和溶剂的相互作用),计算其在结合态与自由态之间的自由能差。近年来,BindFlow等自动化流程的出现显著降低了ABFE的使用门槛——该工具基于GROMACS引擎,内置支持GAFF、OpenFF等多种小分子力场,并在139个配体-靶点对上完成了系统验证,其预测与实验的吻合度接近领域内的金标准方法。
ABFE的优势在于 不依赖任何参考配体 ,能够直接给出单个分子的结合自由能估算,且具有明确的统计力学基础。但其计算成本较高(通常需要数十到数百纳秒的增强采样模拟),且对初始结构的质量敏感。对于D-肽等非标准体系,还需要特别注意力场参数是否覆盖了非天然手性中心的能量描述。ABFE尤其适用于以下场景:针对全新靶点设计首条短肽、评估现有设计是否具备纳摩尔级结合潜力,或验证AI预测结构是否在热力学上可行。
③ 相对自由能微扰计算(FEP)——有阳性分子时的精准优化
在药物发现中最常见也最实用的场景是:已经有一个已知活性的阳性分子(或初始命中分子),需要评估一系列修饰改造后亲和力的提升或下降。此时, 相对自由能微扰(Relative Binding Free Energy, RBFE/FEP) 是最优选择。FEP基于热力学循环原理,通过模拟两个结构相似的分子在结合态和自由态之间的自由能差,直接计算相对结合自由能(ΔΔG)。这种方法的最大优势在于 误差抵消 ——由于分子A和分子B在结构上高度相似,计算过程中系统误差倾向于相互抵消,因此FEP的预测精度通常显著优于MMPBSA和ABFE。
在实践中,FEP要求配体对之间的化学变化较小(如一个官能团的替换或甲基的添加),且能够构建合理的“炼金术路径”从一个分子逐步“变形”为另一个。当存在多个待测分子时,通常以阳性分子为参考节点,构建星形拓扑或线性路径进行所有配体的相对排序。FEP已在工业药物发现中被广泛应用,Schrödinger的FEP+以及开源QligFEP等工具均提供了成熟的实现方案。对于D-肽的优化场景——例如在保持结合模式的前提下逐位点替换D-氨基酸或调整侧链修饰——FEP是量化改造效果的理想工具。
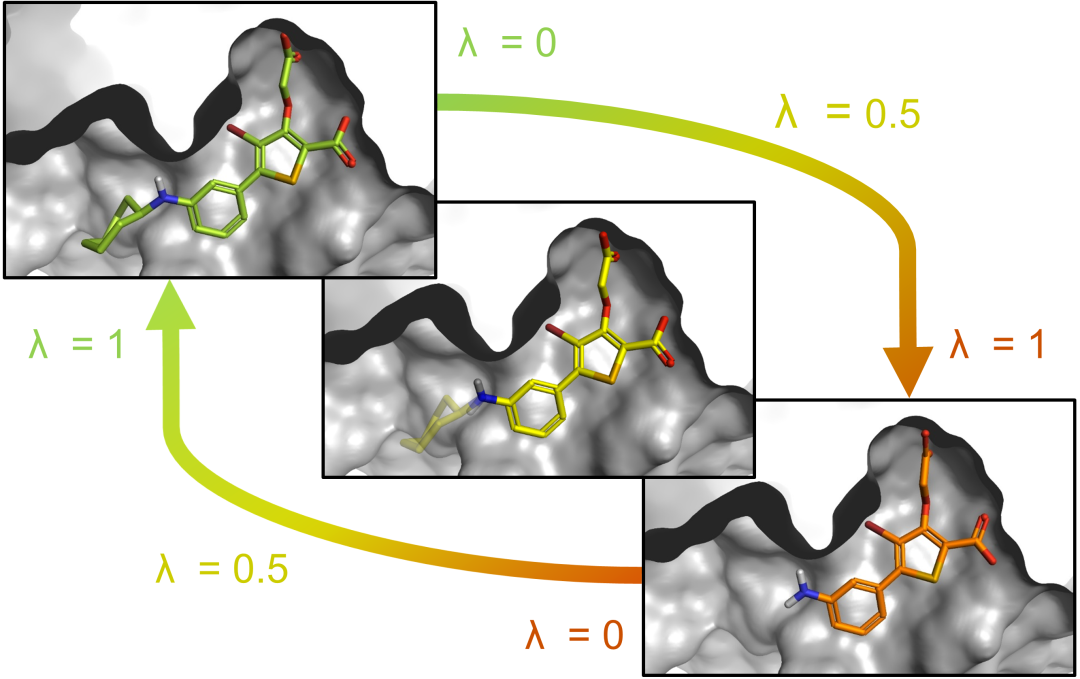
图4. FEP相对自由能计算示意图
策略选择与注意事项
上述三种方法在计算成本、精度和适用场景上形成互补:深度学习亲和力预测最适合超高通量初筛(秒级/千条);ABFE适用于无参考配体的绝对亲和力估算(天级/单分子);FEP则是在有阳性分子时的黄金标准(小时至天级/配体对)。研究者应根据自己的研究阶段和资源条件做出合理选择。无论采用哪种方法,都建议与实验数据建立闭环验证:对于有阳性分子的体系,应首先用该分子校准计算方法(确保FEP或ABFE能复现实验趋势),再推广至待测分子。同时,所有能量计算方法都受限于力场的准确性和采样充分性,其输出应始终作为辅助决策依据,而非实验替代。
4. 深度分析 :如何评估你的短肽设计?
获得稳定的复合物结构后,可通过多维分析量化设计质量:
A. 界面接触分析
利用分析脚本自动生成靶点与短肽之间的距离图和接触图。距离图可直观展示结合口袋的紧凑程度;接触图则帮助识别关键的“热点”残基。
B. 动力学稳定性分析
-
RMSD :评估模拟过程中复合物的整体波动。平稳的曲线意味着结合模式稳定。
-
RMSF :识别短肽中哪些残基较为灵活,哪些残基牢固锚定在结合口袋中。
C. 结合自由能计算 (MMPBSA)
基于AmberTools的MMPBSA.py模块,可定量计算预测结合模式的自由能。这是评估短肽亲和力的关键指标,能够有效区分不同设计序列的优劣。
5. 局限性与重要注意事项(请务必反复阅读)
尽管上述流程集成了当前最先进的结构预测和分子模拟技术,但我们必须坦诚地指出: 没有任何一种计算模型或 自动化流程 能够在工业药物发现场景中做到通用且强健 。以下局限性和注意事项需要研究者在实践中时刻铭记:
建模可信度评估
-
AI预测的物理偏差 :如前所述,共折叠模型可能过度记忆训练数据中的结合模式,而忽略真实的物理化学约束。因此, 不能仅凭AI输出的高置信度打分就判定设计成功 。建议总是对同一体系进行多次重复预测(至少3-5个随机种子),观察结合模式的收敛性。若不同预测给出差异巨大的构象,则需谨慎对待。
-
分子动力学模拟 的力场局限性 :经典力场(如ff14SB、GAFF2)无法描述化学反应、电子极化效应和金属离子配位等复杂行为。对于含有金属离子、共价抑制剂或高度极化基团的体系,可能需要采用QM/MM或极化力场进行补充验证。
-
采样不足问题 :常规的10-100 ns模拟时间可能无法完全采样短肽的所有构象变化,尤其对于高度柔性肽或存在缓慢构象转变的体系。若观察到RMSD持续漂移或结合模式不稳定,应延长模拟时间(至少200-500 ns)或采用增强采样方法(如副本交换、元动力学)。
-
修饰氨基酸参数的质量 :使用GAFF2生成非标准残基参数时,电荷计算方法(如AM1-BCC、RESP)和原子类型划分直接影响能量计算的可靠性。 务必对生成的参数进行目视检查 :键长、键角是否合理?电荷分布是否符合化学直觉?必要时可与DFT计算的静电势进行对比验证。
-
对专家知识的严重依赖 :整个流程中的每一个中间节点——从初始SMILES生成、AI预测结构选择,到结构纠正的质量、模拟参数的设定、轨迹分析的解读——都需要研究者的专业判断。 没有“一键生成”的捷径 。建议团队中至少有一名具有分子建模和模拟经验的成员,负责关键节点的质量控制和异常识别。
-
阳性分子作为“定海神针” :如果针对同一靶点存在已知活性的阳性分子(例如已报道的活性短肽或小分子抑制剂), 强烈建议对该阳性分子执行完全相同的计算流程 。以其结合模式(是否重现实验构象)、稳定性(RMSD曲线是否平稳)和能量(ΔG是否与活性趋势一致)作为参考基准。只有当待测设计的计算结果显著优于或等价于阳性分子,且结合模式合理时,才具有进一步实验验证的价值。反之,若阳性分子本身在计算中表现不佳(例如预测构象与实验差异大、模拟中脱落),则说明当前计算设置存在问题,需要重新优化参数或更换模拟方法。
-
实验反馈是最终裁判 :计算模型的输出永远只是 假说 。任何有前景的短肽设计都应当通过实验(如SPR、ITC、荧光偏振、细胞活性测定)进行验证。计算与实验的迭代优化,才是药物设计的正道。
能量计算方法的适用边界与误用风险
必须承认,当前没有任何一种能量计算方法能够在所有场景下稳定输出可靠的亲和力排序。深度学习亲和力预测模型的最大风险在于训练集偏差——当设计的短肽在序列或结构上与训练数据分布存在差异时(如引入D-氨基酸、非天然残基或环状结构),模型的预测结果可能完全不可靠。ABFE和FEP方法虽然具有更坚实的统计力学基础,但其精度高度依赖于模拟时间是否足够(采样不充分会导致自由能估算偏差)、力场参数是否准确(尤其是对于非标准残基的电荷和二面角参数),以及是否存在未被采样的隐性构象变化。此外,FEP方法要求配体对之间具有明确的化学相似性(通常Tanimoto相似系数>0.7),否则“炼金术路径”将失去物理意义。研究者应充分理解这些方法各自的假设条件和失效模式,避免将计算结果过度解读为“实验测定的亲和力”。当不同方法给出的结论不一致时,优先采用物理基础更扎实的方法(FEP > ABFE > MM(PB)GBSA > 深度学习),但仍需以实验验证为最终标准。
6. 总结
本文介绍的这套 AI预测 + 结构映射 + MD 模拟 工作流,旨在解决D-氨基酸短肽药物设计中的核心技术瓶颈——从手性SMILES生成、AI结构预测,到格式纠正、修饰基团参数构建与物理模拟验证。该流程既利用了AI的高通量筛选能力,又通过基于经典力场的分子动力学模拟确保了物理合理性,同时强调了对阳性对照和专家知识的依赖。
然而,我们再次强调: 计算永远是辅助,实验才是标准 。在短肽药物研发快速发展的今天,工具的革新不断拓展设计的边界,但只有将谨慎的计算与扎实的实验相结合,才能走得更远。欢迎在和我们深入讨论,开启您的D-肽设计之旅。
