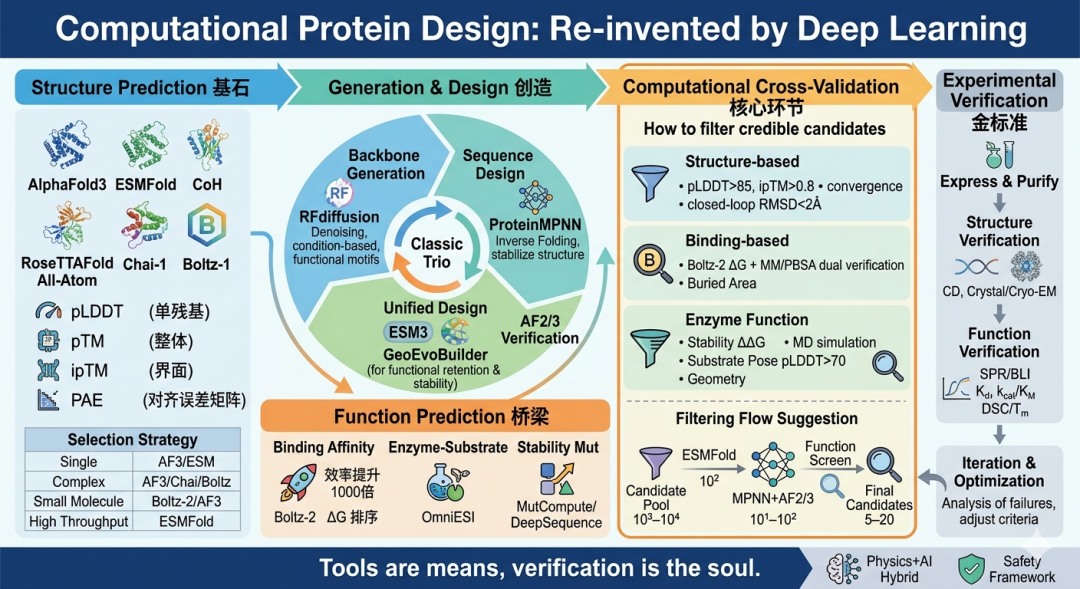
 Figure 1. Panorama of protein design
Figure 1. Panorama of protein design
1. Protein structure prediction: Which "ruler" is the most accurate?
Protein structure prediction is the cornerstone of design. Whether it is to verify the folding of a de novo designed protein or to evaluate whether the designed sequence can form the expected three-dimensional conformation, reliable structure prediction tools are needed.1.1 AlphaFold3: All-around player, but not everything
AlphaFold3 is currently the most comprehensive biomolecule structure prediction model that can handle complexes of proteins, nucleic acids, small molecules and ions. Its core confidence indicators include: pLDDT (predicted Local Distance Difference Test) : Single residue confidence, 0–100, the higher the value, the more credible the local structure is. It is generally considered that >90 is extremely high confidence, 70-90 is credible, 50-70 is low confidence, and <50 is an untrustworthy area. pTM (predicted TM-score) : Confidence of the overall structure, >0.5 indicates that the prediction of the overall folding may be correct ipTM (interface pTM) : Interface confidence, specifically assessing the credibility of interchain interactions in a complex PAE (Predicted Aligned Error) : Predicted alignment error matrix, used to determine the uncertainty in the relative positions of two residues CASP16 evaluation shows that AlphaFold3 is close to experimental accuracy in predicting monomeric protein structures, but for truly novel or non-homologous sequence proteins, predictions still need to be cautious. In terms of protein complex prediction, about 50% of targets are only partially well predicted, and more than 30% are extremely difficult. Atomic-level precision of antibody-antigen complexes and novel protein-protein interfaces remains a major challenge. ⚠️ Usage reminder: AlphaFold3’s ligand binding affinity prediction is not yet reliable and cannot replace specialized binding free energy calculations. Figure 2. AlphaFold online server (https://alphafoldserver.com/)
Figure 2. AlphaFold online server (https://alphafoldserver.com/)
1.2 ESMFold: A speed-first “fast screener”
ESMFold is based on the protein language model and does not rely on multiple sequence alignment (MSA). The inference speed is extremely fast—predictions can be completed in seconds, making it suitable for high-throughput screening scenarios. However, its accuracy is slightly lower than the AlphaFold series on difficult targets, and it is more suitable for preliminary filtering rather than final structure confirmation.1.3 RoseTTAFold All-Atom/Chai-1/Boltz-1: Biomolecular Complex Expertise
These models focus on structure prediction of biomolecular complexes. Among them, Boltz-1 introduces controllability features such as experimental method condition control, distance constraints, and multiple chain template integration, and has unique advantages in complex modeling. It is worth noting that models such as RoseTTAFold All-Atoms, Chai and Boltz have not participated in CASP16 and lack strict third-party evaluation. When selecting, they need to be combined with their own data for internal verification.1.4 Structural prediction tool selection strategy
|
scene |
Recommended tools |
reason |
|
monomeric protein |
AlphaFold3/ESMFold |
The highest accuracy, ESMFold is suitable for rapid preliminary screening |
|
protein-protein complex |
AlphaFold3/Chai-1/Boltz-1 |
Multi-tool cross-validation is the most reliable |
|
protein-small molecule complex |
Boltz-2/AlphaFold3 |
Boltz-2 is specially optimized for combined prediction |
|
High-throughput structure screening modeling |
ESMFold |
Fast, suitable for large-scale structural modeling and prediction |
2. Protein generation and design: from “reading structure” to “writing sequence”
If structure prediction is “reading,” then protein design is “writing”—creating proteins with targeted structure and function from scratch. The two main pillars of this field are Skeleton generation and sequence design .2.1 RFdiffusion: The diffusion revolution in skeleton generation
RFdiffusion is a skeleton generation tool based on the denoising diffusion probability model, which generates the protein backbone skeleton from the noise through step-by-step "denoising". It supports unconditional generation and conditional generation (such as constrained design based on target epitopes or functional motifs). RFdiffusion is widely used to design protein binding agents de novo. By generating a polyglycine skeleton and combining it with ProteinMPNN to assign sequences, the experimental success rate jumped from less than 1% to 7%–35%. In antibody design, fine-tuned RFdiffusion can achieve atomic-level precision CDR conformation generation. Applicable scenarios : A new protein skeleton needs to be generated; conditional design needs to be based on functional motifs (such as enzyme active sites, binding epitopes). ⚠️ Usage reminder: RFdiffusion consumes a lot of computing resources. When using it on a large scale, the computing power cost needs to be considered, and the design structure is not equal to the final structure and must be cross-validated with other tools. Figure 3. RFdiffusion online server (https://app.tamarind.bio/tools/rfdiffusion)
Figure 3. RFdiffusion online server (https://app.tamarind.bio/tools/rfdiffusion)
2.2 ProteinMPNN: The “Inverse Folding Master” of Sequence Design
ProteinMPNN is a representative of the "reverse folding" model - given a target skeleton structure, an amino acid sequence that can be stably folded into the structure is generated. It forms a classic closed loop of "skeleton generation → sequence filling → structure verification" with RFdiffusion. In the actual workflow, the skeleton generated by RFdiffusion will be input into ProteinMPNN to generate multiple candidate sequences (usually 200), and then verified by AlphaFold2/3 folding to screen out the candidates with the highest designability. ⚠️ Usage reminder: ProteinMPNN is only designed based on static structure and does not consider protein folding dynamics. A high design score does not guarantee high experimental expression. Figure 4. ProteinMPNN online server (https://app.tamarind.bio/tools/proteinmpnn)
Figure 4. ProteinMPNN online server (https://app.tamarind.bio/tools/proteinmpnn)
2.3 ESM3/GeoEvoBuilder: A new paradigm for sequence-structure joint design
Models such as ESM3 integrate sequence and structural modes and perform joint design in a unified framework, improving the foldability and functional retention of designed proteins. GeoEvoBuilder is a functional protein design model developed by Peking University's Lai Luhua team. By fusing the structure sequence design model with the protein large language model ESM2, it can simultaneously improve the catalytic activity and thermal stability of the protein. In practical applications, an optimized sequence that increases catalytic efficiency by 10–20 times and thermal stability by about 10°C can be obtained through only a single round of design. This is necessary for Improve stability without sacrificing activity The enzyme engineering scenario is particularly important.2.4 Generate tool selection strategy
|
Task |
Recommended combination |
illustrate |
|
Designing protein binders from scratch |
RFdiffusion + ProteinMPNN + AF2/3 |
Classic three-piece set |
|
Optimize natural enzyme activity and stability |
GeoEvoBuilder |
Designed to retain functionality |
|
Antibody/Nanobody Design |
Fine-tuned version of RFdiffusion + ProteinMPNN |
Requires fine-tuning using antibody complex data |
|
Only sequence optimization is required |
ProteinMPNN/ESM-IF |
It can be used directly when there is a skeleton structure |
3. Function prediction tools: the bridge from structure to function
3.1 Boltz-2: A “game changer” for binding affinity prediction
Boltz-2 is a basic model of structural biology. Its most prominent innovations are: It is the first AI model to approach FEP accuracy in predicting protein-small molecule binding affinity. . Boltz-2 predicts binding affinity (ΔG) directly from 3D geometry without docking scoring or molecular dynamics simulations. What is even more impressive is the efficiency - a single prediction takes about tens of seconds and costs a few cents, while FEP takes 6–12 hours and costs about $100 per prediction, which is an efficiency increase of more than 1,000 times. Applicable scenarios : Rapid assessment of protein-small molecule binding affinity; ΔG ranking in virtual screening. ⚠️ Usage reminder: Boltz-2 does not predict binding sites, model flexible loop regions or conformational dynamics. Its confidence score is not directly equivalent to affinity, so the Boltz-ABFE pipeline (Boltz-2 structure prediction + absolute FEP calculation) is recommended as a more robust evaluation scheme. Figure 5. Boltz2 online server (https://proteiniq.io/app/boltz-2)
Figure 5. Boltz2 online server (https://proteiniq.io/app/boltz-2)
3.2 OmniESI/MutCompute/DeepSequence: Enzyme-substrate interaction and stability prediction
In enzyme engineering, OmniESI predicts enzyme-substrate interactions through two-stage progressive conditional deep learning, which can be used to guide substrate-specific modifications. MutCompute and DeepSequence predict the impact of mutations on protein stability based on multiple sequence alignments and language models, and are suitable for guiding directed evolution and mutation site screening.4. Computational cross-validation: How to select the most credible candidates?
This is the core part of this article. The quality of screening in the calculation stage directly determines the success rate of experimental verification. . The results of a single model should not be delivered directly to experiments and must undergo multi-dimensional cross-validation.4.1 Three-level verification of structural design
The first level: Confidence index (quick preliminary screening)- pLDDT: Confidence distribution of structure predictions. Screen candidates with pLDDT >85, focusing on local pLDDT values in key functional regions (e.g., binding interface, catalytic residues)
- pTM/ipTM: ipTM>0.8 indicates that the interface prediction is highly credible
- PAE: PAE_interaction<7.5 Å is the empirical threshold for judging the reliability of interface interaction
4.2 Energy verification combined with class design
Boltz-2 ΔG +MM/PBSA double check Although Boltz-2 is fast, its predictions are not infallible. A more robust approach is: ΔG ranking of candidate complexes using Boltz-2 to screen out the 10%–20% with the highest predicted affinity Run MM/PBSA (Molecular Mechanics/Poisson–Boltzmann Surface Area) calculations on shortlist candidates and cross-check binding free energies When possible, run FEP/ABFE (such as the Boltz-ABFE pipeline) on the best candidate to obtain a more accurate binding free energy estimate Extra checks : Calculate the buried surface area (Buried Surface Area) and the number of interface hydrogen bonds/hydrophobic interactions of the complex, compare them with the natural binding interface, and eliminate candidates with weak interface interactions or geometric abnormalities. Based on the understanding of the system, the rationality of key binding sites is confirmed through expert knowledge.4.3 Multidimensional verification of enzyme functional design
Enzyme design requires comprehensive evaluation of three dimensions: Structural stability, substrate binding paradigm, catalytic activity . Stability verification : Use ESM-1v/MutCompute/DeepSequence to predict the impact of mutations on folding free energy (ΔΔG), and screen mutations with ΔΔG≤0 kcal/mol (i.e. mutations that do not destroy stability) Run short-term molecular dynamics simulations (e.g. 10 ns) to assess overall structural stability and calculate backbone RMSF, focusing on whether the flexibility of residues near the active site is appropriate. Substrate binding paradigm validation :- Use AlphaFold3/Boltz-2 to predict the structure of the enzyme-substrate complex and calculate the confidence of the ligand docking pose (pLDDT ligand >70 is the recommended threshold)
- Evaluate whether the geometric relationship (e.g. distance, angle) between catalytic residues and substrate is consistent with known catalytic mechanisms
- Use Boltz-2 to evaluate substrate binding ΔG to ensure that designed mutations do not impair substrate binding ability
4.4 Recommendations for comprehensive screening process
Recommended screening process (from rough to fine) :- generation phase : RFdiffusion/GeoEvoBuilder generates candidate pool (order of magnitude: 10³–10⁴)
- Preliminary screening of structure : ESMFold quickly folds all candidates, and pLDDT<70 is directly eliminated (the order of magnitude is reduced to 10²)
- Design closed loop : ProteinMPNN + AF2/3 closed-loop verification, elimination of RMSD>2 Å (order of magnitude reduced to 10¹–10²)
5. Experimental verification: the end point of calculation and the starting point of truth
No matter how finely calculated the filtering is, Experimental validation remains the ultimate gold standard .5.1 Recommended experimental verification process
Expression and purification : Carry out gene synthesis, expression and purification of candidate sequences selected by calculation. A high design score does not guarantee high expression, this is the first "experimental level". Structural verification :-
Circular dichroism spectroscopy (CD) confirms secondary structure composition
-
Analyze the crystal structure or cryo-electron microscopy structure when conditions permit, and compare it with the computational model
-
Combined with design : Surface plasmon resonance (SPR) or biolayer interference (BLI) determines Kd. Existing data show that about 19% of the binders designed by RFdiffusion+ProteinMPNN show binding signals at a concentration of 10 μM, but the proportion reaching high affinity is much lower - which means that even after computational screening, tens to hundreds of candidates still need to be experimentally tested.
-
enzyme design : Determine kcat/KM to evaluate catalytic efficiency; thermal stability is determined by differential scanning calorimetry (DSC) or thermal drift method Tm
-
Stability verification : Chemical denaturation or thermal denaturation experiments to evaluate ΔG fold
5.2 Efficiency improvement of calculation-experiment closed loop
A well-designed computational-experimental closed loop can shorten the "sequence → functional protein" cycle from months to weeks, but this requires close collaboration between the computational and experimental teams. The computing side delivers not only a set of sequences, but also the prediction confidence of each sequence, potential risk tips, and recommended experiment prioritization. , helping the experimental side allocate resources efficiently.Conclusion: Tools are just means, verification is the soul
Deep learning tools for protein design are evolving at a dizzying speed - from AlphaFold to AlphaFold3, from Boltz-1 to Boltz-2, model architectures continue to break through, and performance boundaries continue to extend. The 2024 Nobel Prize in Chemistry will be awarded to AlphaFold and the pioneers of protein design, marking the transition of this field from "frontier exploration" to "mainstream paradigm". But beyond the exciting tool iterations, one simple truth always holds true: Calculation results that have not been fully cross-validated should not enter the experimental stage. Multi-tool comparison, confidence assessment, energy verification, closed-loop verification—these seemingly cumbersome calculation steps are exactly the key to the success or failure of the design. Looking forward to the future, we expect to see the emergence of more “physics + AI” hybrid frameworks, more robust security frameworks to address dual-use risks, and closer computing-experiment integration platforms. At present, mastering the wisdom of tool selection and establishing a rigorous calculation verification system are the internal skills that every protein designer needs to cultivate most.Follow us and witness a new era of computational changes in protein research and development!
Contact information: information@yunfeidu.com
👇 Click on the business card below to follow "Yunfeidu Technology"
