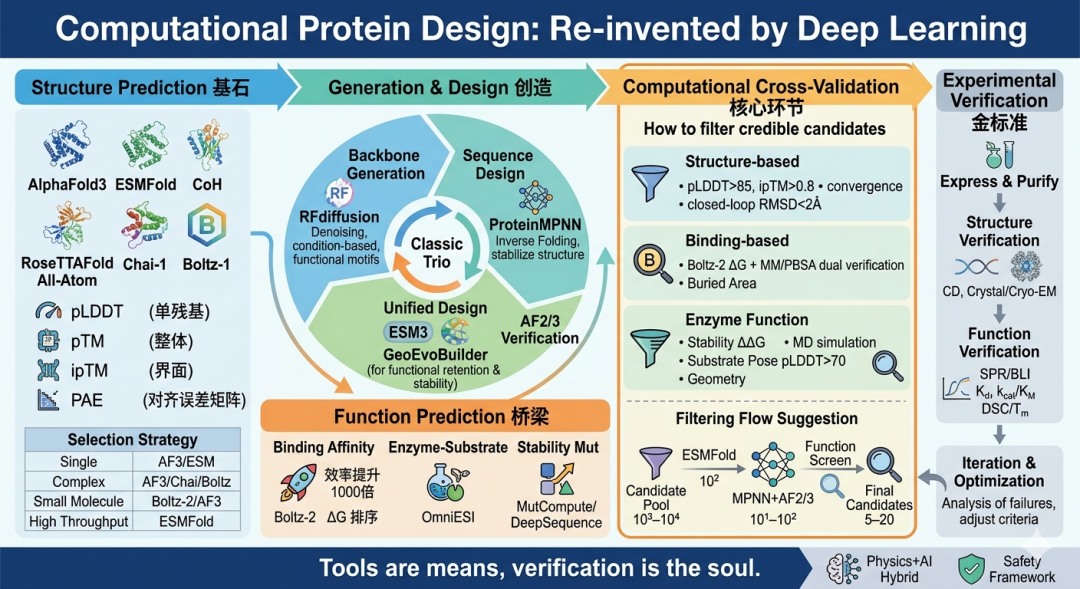
 图1.蛋白质设计全景
图1.蛋白质设计全景
一、蛋白质结构预测:哪把“尺子”最准?
蛋白质结构预测是设计的基石。无论是验证从头设计蛋白的折叠情况,还是评估设计序列能否形成预期的三维构象,都需要可靠的结构预测工具。1.1 AlphaFold3:全能型选手,但非万能
AlphaFold3是目前最全面的生物分子结构预测模型,可处理蛋白质、核酸、小分子和离子的复合物。其核心置信度指标包括: pLDDT(predicted Local Distance Difference Test) :单残基置信度,0–100,值越高局部结构越可信。一般认为>90为极高置信度,70–90为可信,50–70为较低置信度,<50为不可信区域 pTM(predicted TM-score) :整体结构置信度,>0.5表示预测整体折叠可能正确 ipTM(interface pTM) :界面置信度,专门评估复合物中链间相互作用的可信度 PAE(Predicted Aligned Error) :预测的对齐误差矩阵,用于判断两个残基相对位置的不确定性 CASP16评估显示,AlphaFold3在单体蛋白质结构预测上已接近实验精度,但对于真正新颖或无同源序列的蛋白质,预测仍需谨慎。在蛋白质复合物预测上,约50%的靶点仅获得部分良好预测,超过30%极具难度。抗体-抗原复合物和新型蛋白-蛋白界面的原子级精度仍是重大挑战。 ⚠️ 使用提醒:AlphaFold3的配体结合亲和力预测尚不可靠,不能替代专门的结合自由能计算。 图2. AlphaFold在线服务器(https://alphafoldserver.com/)
图2. AlphaFold在线服务器(https://alphafoldserver.com/)
1.2 ESMFold:速度优先的“快速筛查器”
ESMFold基于蛋白质语言模型,不依赖多序列比对(MSA),推理速度极快——可在秒级完成预测,适合高通量筛选场景。但其精度在高难度靶点上略低于AlphaFold系列,更适合用于初步过滤而非最终结构确认。1.3 RoseTTAFold All-Atom / Chai-1 / Boltz-1:生物分子复合物专长
这些模型专注于生物分子复合物的结构预测。其中Boltz-1引入了实验方法条件控制、距离约束和多重链模板整合等可控性特征,在复合物建模上有独特优势。值得关注的是,RoseTTAFold All-Atoms、Chai和Boltz等模型未参与CASP16,缺乏严格的第三方评估,选用时需要结合自身数据做内部验证。1.4 结构预测工具选择策略
|
场景 |
推荐工具 |
理由 |
|
单体蛋白质 |
AlphaFold3 / ESMFold |
精度最高,ESMFold适合快速初筛 |
|
蛋白-蛋白复合物 |
AlphaFold3 / Chai-1 / Boltz-1 |
多工具交叉验证最稳妥 |
|
蛋白-小分子复合物 |
Boltz-2 / AlphaFold3 |
Boltz-2在结合预测上有专门优化 |
|
高通量结构筛选建模 |
ESMFold |
速度快,适合大规模结构建模和预测 |
二、蛋白质生成与设计:从“读结构”到“写序列”
如果说结构预测是“读”,那么蛋白质设计就是“写”——从零创造具有目标结构和功能的蛋白质。这一领域的两大支柱是 骨架生成 和 序列设计 。2.1 RFdiffusion:骨架生成的扩散革命
RFdiffusion是基于去噪扩散概率模型的骨架生成工具,通过逐步“去噪”从噪声中生成蛋白质主链骨架。它支持无条件生成和条件生成(如基于目标表位或功能基序的约束设计)。 RFdiffusion被广泛应用于从头设计蛋白结合剂。通过生成多聚甘氨酸骨架,再结合ProteinMPNN赋予序列,实验成功率从不足1%跃升至7%–35%。在抗体设计中,经过微调的RFdiffusion可实现原子级精度的CDR构象生成。 适用场景 :需要生成全新蛋白质骨架;需要基于功能基序(如酶活性位点、结合表位)进行条件设计。 ⚠️ 使用提醒:RFdiffusion计算资源消耗较大,大规模使用时需考虑算力成本,且设计结构不等于最终结构,须通过其他工具交叉验证。 图3. RFdiffusion在线服务器(https://app.tamarind.bio/tools/rfdiffusion)
图3. RFdiffusion在线服务器(https://app.tamarind.bio/tools/rfdiffusion)
2.2 ProteinMPNN:序列设计的“逆折叠大师”
ProteinMPNN是“逆折叠”模型的代表——给定目标骨架结构,生成能够稳定折叠为该结构的氨基酸序列。它与RFdiffusion形成经典的“骨架生成→序列填充→结构验证”闭环。 在实际工作流中,RFdiffusion生成的骨架会输入ProteinMPNN产生多条候选序列(通常200条),再经AlphaFold2/3折叠验证,筛选出设计性(designability)最高的候选。 ⚠️ 使用提醒:ProteinMPNN仅根据静态结构进行设计,不考虑蛋白质折叠动力学,高设计性评分不保证实验高表达。 图4. ProteinMPNN在线服务器(https://app.tamarind.bio/tools/proteinmpnn)
图4. ProteinMPNN在线服务器(https://app.tamarind.bio/tools/proteinmpnn)
2.3 ESM3 / GeoEvoBuilder:序列-结构联合设计的新范式
ESM3等模型整合了序列与结构模态,在统一框架中进行联合设计,提升了设计蛋白的可折叠性和功能保留度。 GeoEvoBuilder是北大来鲁华团队开发的功能蛋白设计模型,通过将结构序列设计模型与蛋白质大语言模型ESM2融合,能同时提升蛋白的催化活性和热稳定性。实际应用中,仅通过单轮设计即获得催化效率提升10–20倍、热稳定性提高约10℃的优化序列。这对于需要在 不牺牲活性的前提下提升稳定性 的酶工程场景尤为重要。2.4 生成工具选择策略
|
任务 |
推荐组合 |
说明 |
|
从头设计蛋白结合剂 |
RFdiffusion + ProteinMPNN + AF2/3 |
经典三件套 |
|
优化天然酶活性与稳定性 |
GeoEvoBuilder |
专为功能保留设计 |
|
抗体/纳米抗体设计 |
微调版RFdiffusion + ProteinMPNN |
需使用抗体复合物数据微调 |
|
仅需序列优化 |
ProteinMPNN / ESM-IF |
有骨架结构时可直接使用 |
三、功能预测工具:从结构到功能的桥梁
3.1 Boltz-2:结合亲和力预测的“游戏规则改变者”
Boltz-2是一个结构生物学基础模型,其最突出的创新在于: 它是首个在蛋白质-小分子结合亲和力预测上接近FEP精度的AI模型 。Boltz-2直接从3D几何预测结合亲和力(ΔG),无需对接打分,也无需分子动力学模拟。 更令人印象深刻的是效率——单次预测约几十秒、成本几美分,而FEP需要6–12小时、成本约100美元/预测,效率提升1000倍以上。 适用场景 :蛋白质-小分子结合亲和力快速评估;虚拟筛选中的ΔG排序。 ⚠️ 使用提醒:Boltz-2不预测结合位点,不建模柔性环区或构象动力学。其置信度评分不直接等同于亲和力,因此Boltz-ABFE管线(Boltz-2结构预测 + 绝对FEP计算)被推荐作为更稳健的评估方案。 图5. Boltz2 在线服务器(https://proteiniq.io/app/boltz-2)
图5. Boltz2 在线服务器(https://proteiniq.io/app/boltz-2)
3.2 OmniESI / MutCompute / DeepSequence:酶-底物互作与稳定性预测
在酶工程中,OmniESI通过两阶段渐进式条件深度学习预测酶-底物相互作用,可用于指导底物特异性改造。MutCompute和DeepSequence则基于多序列比对和语言模型预测突变对蛋白稳定性的影响,适用于指导定向进化和突变位点筛选。四、计算交叉验证:如何筛选出最可信的候选?
这是本文最核心的部分。 计算阶段的筛选质量直接决定实验验证的成功率 。单一模型的结果不应直接交付实验,必须经过多维度交叉验证。4.1 结构类设计的三层验证
第一层:置信度指标(快速初筛)- pLDDT:结构预测的置信度分布。筛选pLDDT>85的候选,重点关注关键功能区域(如结合界面、催化残基)的局部pLDDT值
- pTM / ipTM:ipTM>0.8表示界面预测高度可信
- PAE:PAE_interaction<7.5 Å是判断界面互作可靠性的经验阈值
4.2 结合类设计的能量验证
Boltz-2 ΔG +MM/PBSA 双重校验 Boltz-2虽然速度快,但其预测并非绝对可靠。更稳健的做法是: 用Boltz-2对候选复合物进行ΔG排序,筛选出预测亲和力最高的10%–20% 对短名单候选运行MM/PBSA(Molecular Mechanics/Poisson–Boltzmann Surface Area)计算,交叉校验结合自由能 条件允许时,对最优候选运行FEP/ABFE(如Boltz-ABFE管线)获得更精确的结合自由能估计 额外检查 :计算复合物的埋藏表面积(Buried Surface Area)和界面氢键/疏水相互作用数量,与天然结合界面做比对,剔除界面相互作用过弱或几何异常的候选。根据对体系的认知,通过专家知识确认关键结合位点的合理性。4.3 酶功能设计的多维验证
酶设计需综合评估三个维度: 结构稳定性、底物结合范式、催化活性 。 稳定性验证 : 用ESM-1v / MutCompute / DeepSequence预测突变对折叠自由能(ΔΔG)的影响,筛选ΔΔG≤0 kcal/mol的突变(即不破坏稳定性的突变) 运行短时分子动力学模拟(如10 ns),评估整体结构稳定性,计算骨架RMSF,重点关注活性位点附近残基的柔性是否合适 底物结合范式验证 :- 用AlphaFold3/Boltz-2预测酶-底物复合物结构,计算配体对接pose的置信度(pLDDT配体>70为推荐阈值)
- 评估催化残基与底物的几何关系(如距离、角度)是否符合已知的催化机制
- 用Boltz-2评估底物结合ΔG,确保设计突变未削弱底物结合能力
4.4 综合筛选流程建议
推荐筛选流程(由粗到精) :- 生成阶段 :RFdiffusion/GeoEvoBuilder生成候选池(数量级:10³–10⁴)
- 结构初筛 :ESMFold快速折叠所有候选,pLDDT<70直接剔除(数量级降至10²)
- 设计性闭环 :ProteinMPNN + AF2/3闭环验证,RMSD>2 Å的剔除(数量级降至10¹–10²)
五、实验验证:计算的终点,真理的起点
无论计算筛选多么精细, 实验验证仍然是最终的金标准 。5.1 推荐的实验验证流程
表达与纯化 :将计算筛选出的候选序列进行基因合成、表达和纯化。高设计性评分不保证高表达,这是第一个“实验关卡”。 结构验证 :-
圆二色谱(CD)确认二级结构组成
-
条件允许时解析晶体结构或冷冻电镜结构,与计算模型比对
-
结合设计 :表面等离子体共振(SPR)或生物层干涉(BLI)测定Kd。现有数据显示,RFdiffusion+ProteinMPNN设计的结合剂中,约19%的候选在10 μM浓度下显示结合信号,但达到高亲和力的比例要低得多——这意味着即使经过计算筛选,仍需要实验测试数十至数百个候选。
-
酶设计 :测定kcat/KM,评估催化效率;热稳定性通过差示扫描量热法(DSC)或热漂移法测定Tm
-
稳定性验证 :化学变性或热变性实验评估ΔG折叠
5.2 计算-实验闭环的效率提升
一个设计完备的计算-实验闭环可将“序列→功能蛋白”的周期从数月缩短至数周,但这需要计算团队与实验团队紧密协作。 计算侧交付的不仅是一组序列,还应包括每一条序列的预测置信度、潜在风险提示和推荐的实验优先级排序 ,帮助实验侧高效分配资源。结语:工具只是手段,验证才是灵魂
蛋白质设计的深度学习工具正在以令人目眩的速度演进——从AlphaFold到AlphaFold3,从Boltz-1到Boltz-2,模型架构不断突破,性能边界持续外延。2024年诺贝尔化学奖授予AlphaFold和蛋白质设计的开拓者,标志着这一领域从“前沿探索”走向“主流范式”。 但在激动人心的工具迭代之外,一个朴素的真理始终成立: 没有经过充分交叉验证的计算结果,不应该进入实验阶段。 多工具比对、置信度评估、能量校验、闭环验证——这些看似繁琐的计算步骤,恰恰是决定设计成败的关键。 面向未来,我们期待看到更多“物理+AI”混合框架的出现、更稳健的安全框架应对双重用途风险,以及更紧密的计算-实验一体化平台。而在当下,掌握工具的选择智慧,建立严谨的计算验证体系,就是每一位蛋白质设计者最需要修炼的内功。关注我们,一起见证计算改变蛋白质研发的新时代!
联系方式:information@yunfeidu.com
👇 点击下方名片,关注“云飞渡科技”
